


Clin. Pract. 2024, 14(3), 729-738; https://doi.org/10.3390/clinpract14030058 (registering DOI) - 25 Apr 2024
Abstract
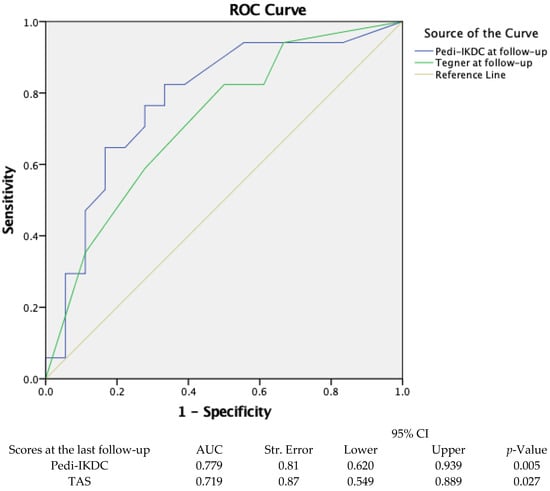
(1) Background: Full-thickness rotator cuff tears (RCTs) impact 25% of those over 60 and 50% over 80; however, minimal data exists on public understanding; (2) Methods: The primary outcome was to determine the public’s baseline understanding of RCTs utilizing a 36-question survey regarding
[...] Read more.
(1) Background: Full-thickness rotator cuff tears (RCTs) impact 25% of those over 60 and 50% over 80; however, minimal data exists on public understanding; (2) Methods: The primary outcome was to determine the public’s baseline understanding of RCTs utilizing a 36-question survey regarding anatomy and function, risk factors, diagnosis and treatment options, and expectations. Secondarily, we evaluated the effect of an educational video and informational handout created by the authors to improve understanding. Participants ≥ 18 years were recruited from the senior author’s clinic and online discussion platforms over a 5-month period; (3) Results: Baseline surveys were completed by 382 individuals: 56% men, 64% Caucasian, 27% with at least a master’s degree, and 56% with very little or no RCT knowledge. Mean correct answer scores improved from 47% to 68% posteducational intervention (p < 0.001). Males, higher education level, healthcare experience, and a higher self-rated understanding of RCTs were significantly correlated with higher survey performance (p < 0.001); (4) Conclusions: The public’s knowledge of RCTs at baseline was poor, with demographic factors correlating with survey performance. The educational intervention effectively enhanced participants’ understanding. By focusing on common misconceptions, this data can help clinicians align patient expectations and enhance patient outcomes.
Full article
(This article belongs to the Special Issue Shoulder Disorders: Diagnosis and Treatment)
►
Show Figures
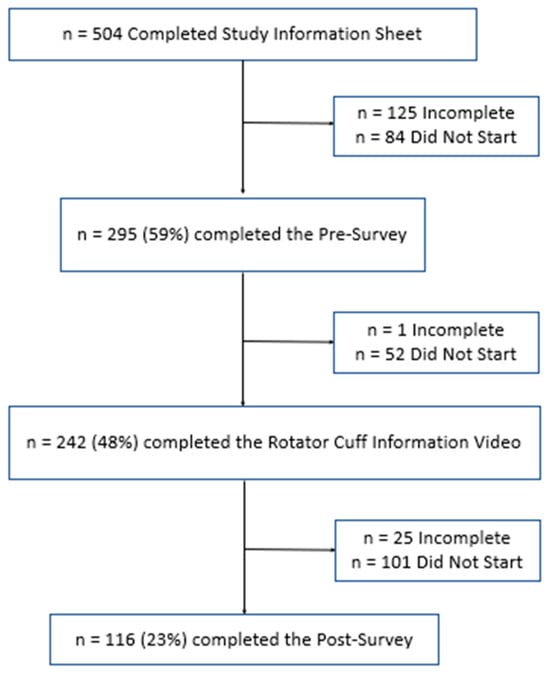
Figure 1



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}