


Clin. Pract. 2024, 14(3), 749-764; https://doi.org/10.3390/clinpract14030060 - 26 Apr 2024
Abstract
Background: Few studies have correlated maternal and neonatal Vit D (25(OH)D) levels at birth in Greece. We investigated this potential association, taking into account the administration or not of low doses (400–800 IU) of prenatal Vit D supplements. Our study contributes evidence not
[...] Read more.
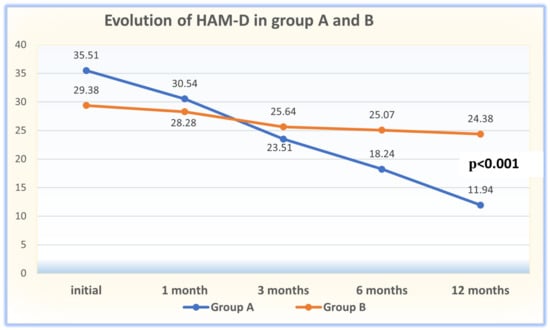
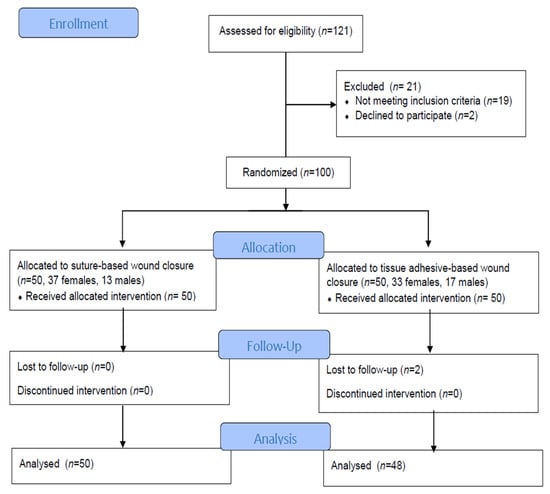
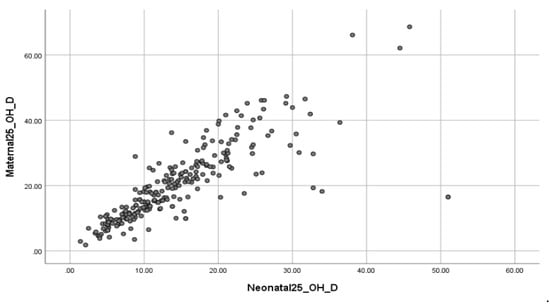
Background: Few studies have correlated maternal and neonatal Vit D (25(OH)D) levels at birth in Greece. We investigated this potential association, taking into account the administration or not of low doses (400–800 IU) of prenatal Vit D supplements. Our study contributes evidence not only to the small amount of existing literature regarding the above correlation, but also to the topic of maternal and neonatal vitamin D deficiency (VDD) during pregnancy in Mediterranean countries, such as Greece. Methods: A cross-sectional study was conducted on 248 neonates and their mothers from September 2019 to January 2022. Blood samples of 25(OH)D were studied at the time of delivery. Frequency counts and percentages were registered, and logistic regression was used to investigate the independent factors associated with maternal Vit D status. The Chi-square test and the Pearson coefficient were used to demonstrate a possible association between maternal and neonatal 25(OH)D levels. Results: Our findings show a high prevalence of VDD in Greek women and their newborns at birth. This was observed not only in women who did not receive Vit D supplements, but also in all the study groups, especially in the autumn and winter months. We observed that mothers who received low doses (400–800 IU) of prenatal Vit D supplements increased both their own 25(OH)D concentrations and those of their newborns; however, the latter did not seem to be completely covered by the prenatal administration of Vit D because, although their 25(OH)D concentrations increased, they never reached sufficient 25(OH)D levels, unlike their mothers who reached sufficient concentrations. Conclusions: Overall, this study highlights the strong association between maternal and neonatal 25(OH)D concentrations at the end of gestation. However, neonates tended to show even lower 25(OH)D concentrations relative to maternal 25(OH)D concentrations. The same phenomenon was observed irrespective of the administration of Vit D supplements during pregnancy. Moreover, this is what was observed concerning the administration of formulations with 400–800 IU of Vit D, which the doctors in our clinic used in the present study. In any case, more clinical studies related to the administration of higher doses of Vit D supplementation to pregnant women would lead to more reliable conclusions. Without a doubt, the measurement of maternal vitamin D status during pregnancy provides opportunities for preventive and therapeutic interventions in the mother–infant pair.
Full article
(This article belongs to the Special Issue 2024 Feature Papers in Clinics and Practice)
►
Show Figures
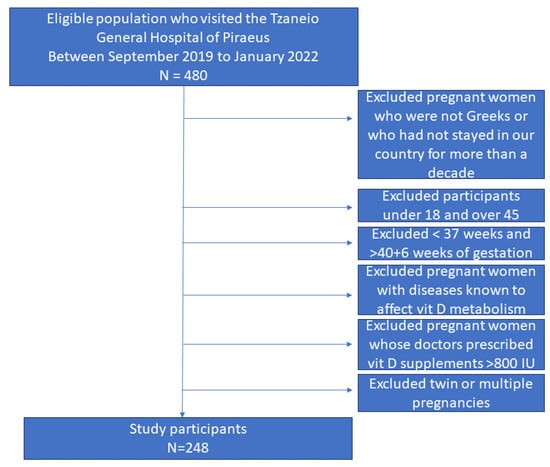
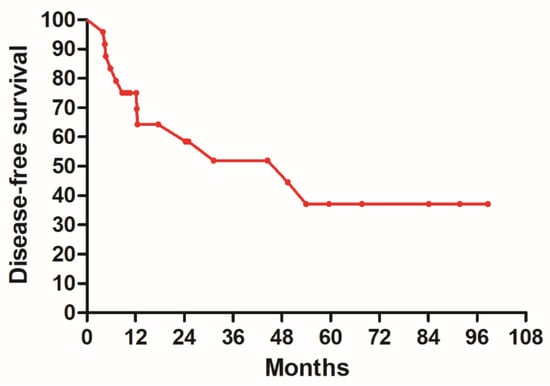
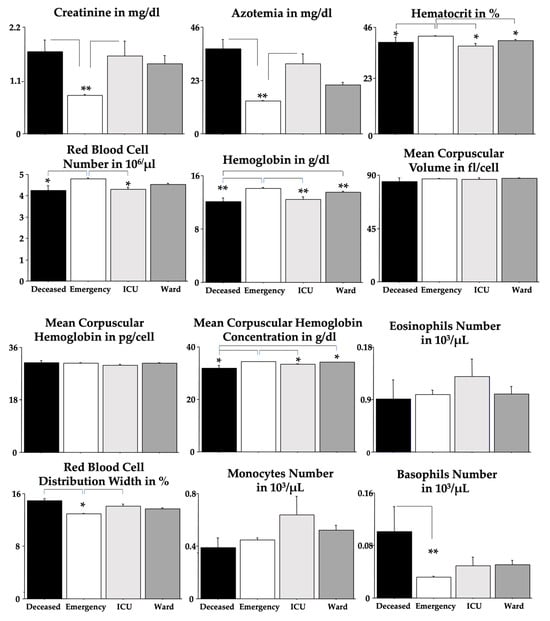
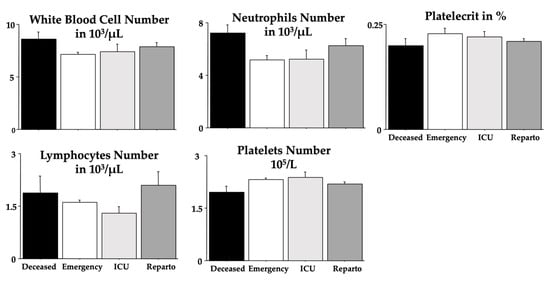
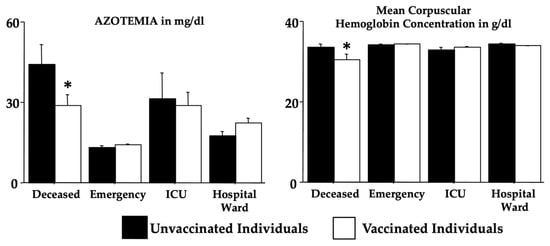
Figure 1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}