


Clin. Pract. 2024, 14(3), 718-728; https://doi.org/10.3390/clinpract14030057 - 24 Apr 2024
Abstract
►
Show Figures
Purpose: To analyse the relationship between the different habits that occur in childhood and the different malocclusions in the three planes of space. Material and methods: A clinical examination of 106 children between 5 and 12 years of age was carried out and
[...] Read more.
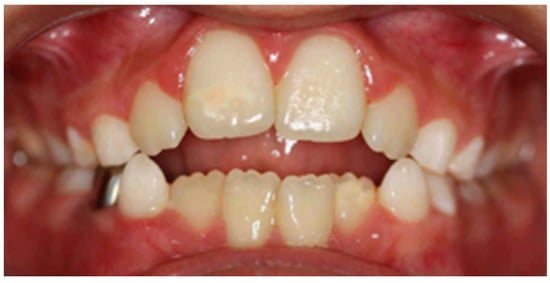
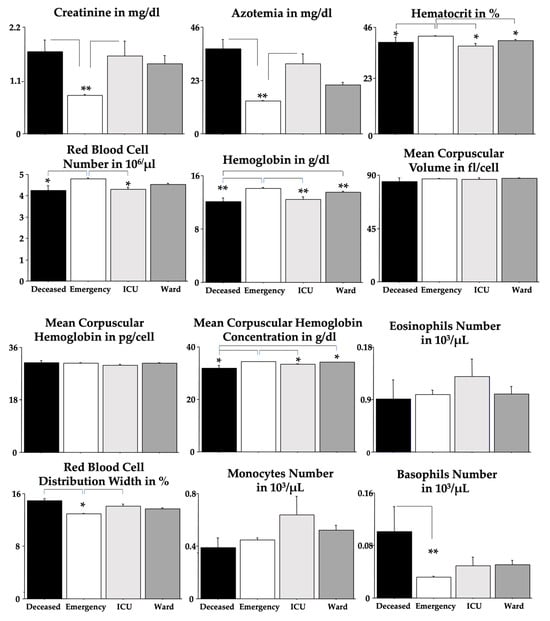
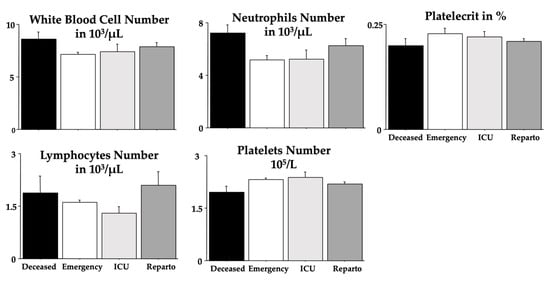
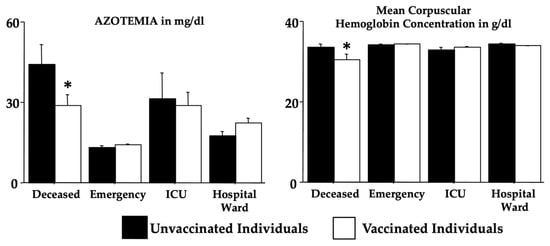
Purpose: To analyse the relationship between the different habits that occur in childhood and the different malocclusions in the three planes of space. Material and methods: A clinical examination of 106 children between 5 and 12 years of age was carried out and a survey validated by professors of the Faculty of Dentistry of the University of Seville was made for the parents in order to identify the habits and relate them to the possible malocclusions detected in the child’s mouth. Results: 72.64% of the sample presented a malocclusion in at least one of the three planes of space, with a similar distribution. When correlating the variables, statistically significant relationships were observed in the vertical plane with atypical swallowing (p = 0 < 0.05; V > 0.3) and lip sucking (p = 0 < 0.05; V > 0.3) and in the horizontal plane with oral breathing (p = 0 < 0.05; V > 0.3), atypical swallowing (p = 0 < 0.05; V < 0.3) and digital sucking (p = 0 < 0.05; V < 0.3). Conclusions: It has been observed that the prevalence and prolongation of habits in childhood is increasing, so it is essential to detect pernicious habits at an early age to prevent the establishment of malocclusions and to favour the correct craniofacial growth of the child.
Full article
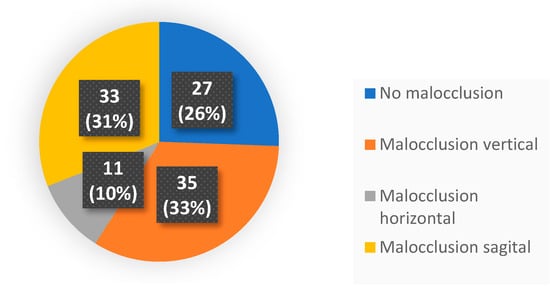
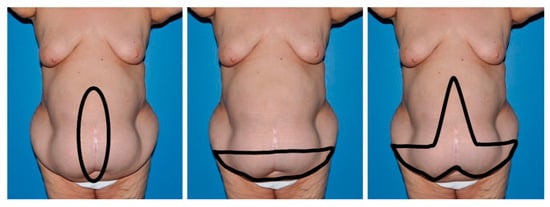
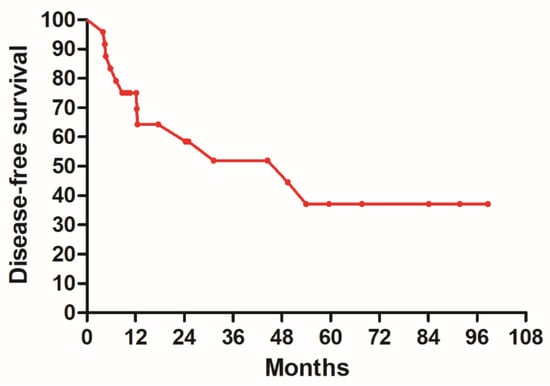
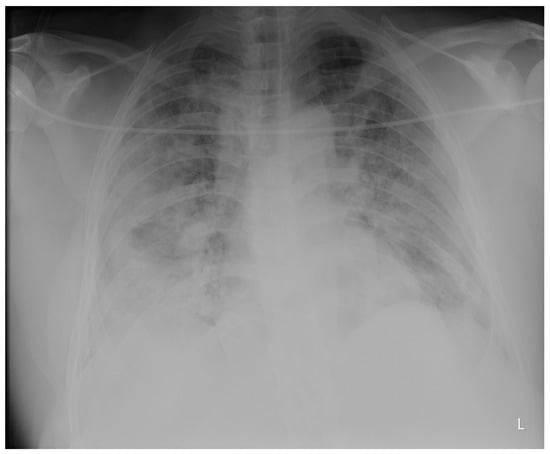
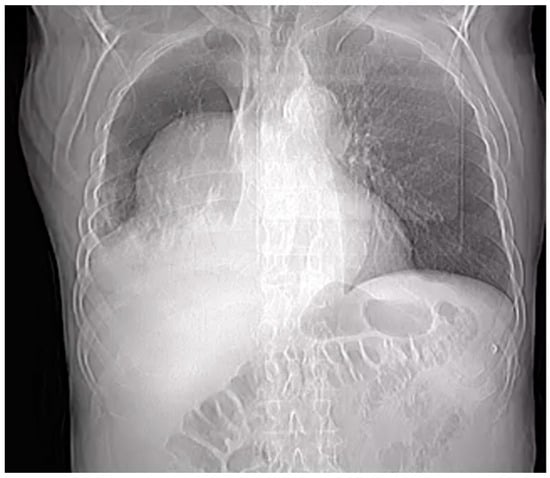
Figure 1



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}